RNA Sequencing
RNA, Sequencing
Article: From RNA-seq reads to differential expression results
Processing high-throughput RNA sequencing data and detecting differential expression
DNA may get most of the public’s attention, but it is gene expression and regulation that orchestrate the dynamics of cell function and physiology. The majority of variants identified in genome-wide association studies (GWAS) occur in noncoding regions of DNA, underscoring the significance of gene expression and regulation in the mechanisms of disease. Sequencing steady-state RNA in a sample, known as RNA-seq, is free from many of the limitations of previous technologies, such as the dependence on prior knowledge of the organism, as required for microarrays and PCR. RNA‑Seq is a powerful sequencing-based method that captures a full and informative spectrum of gene expression data. Most RNA-seq experiments take a sample of purified RNA, shear it, convert it to cDNA ad sequence on a high-throughput platform, such as the illumina GA/ HiSeq, SOLiD or Roche 454.
The following figure outlines the processing pipeline used for detecting differential expression (DE) in RNA-seq.
- Mapping
- The first step in turning millions of short reads into a quantification of expression is the read mapping or alignment. At its simplest, the task of mapping is to find the unique location where a short read is identical to the reference.
- Summarizing mapped reads
- Having obtained genomic locations for as many reads as possible, the next task is to summarize and aggregate reads over some biologically meaningful unit, such as exons, transcripts or genes;
- Normalization
- Normalization enables accurate comparisons of expression levels between and within samples. Because transcripts have higher read counts (at the same expression level), a common method for within-library normalization is to divide the summarized counts by the length of the gene, such as RPKM (reads per kilobase of exon model per million mapped reads)
- Differential expression
- The goal of a differential expression analysis is to highlight genes that have changed significantly in abundance across experimental conditions. In general, this means taking a table of summarized count data for each library and performing statistical testing between samples of interest.
- Systems biology: going beyond gene lists
- Gene expression studies are laying the groundwork for advances in precision medicine by identifying potential therapeutic biomarkers and drug targets.
- There is wide scope for integrating the results of RNA-seq data with other sources of biological data to establish a more complete picture of gene regulation
- RNA-seq has been used in conjunction with genotyping data to identify genetic loci responsible for variation in gene expression between individuals (expression quantitative trait loci or eQTLs)
- integration of expression data with transcription factor binding, RNA interference, histone modification and DNA methylation information has the potential for greater understanding of a variety of regulatory mechanisms.
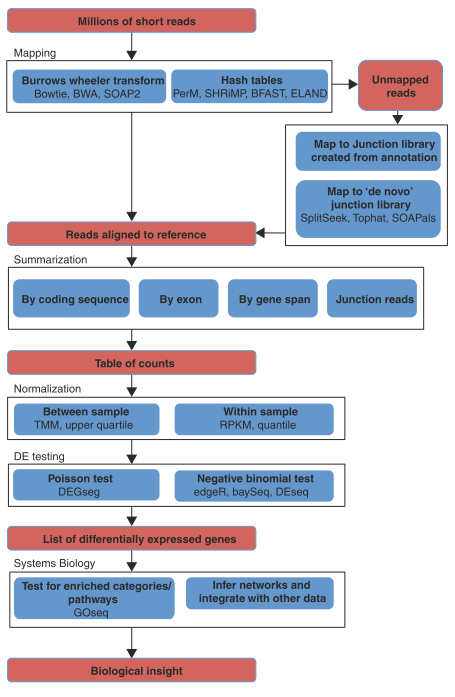
Figure. Overview of the RNA-seq analysis pipeline for detecting differential expression.
Advances in RNA-Seq techniques
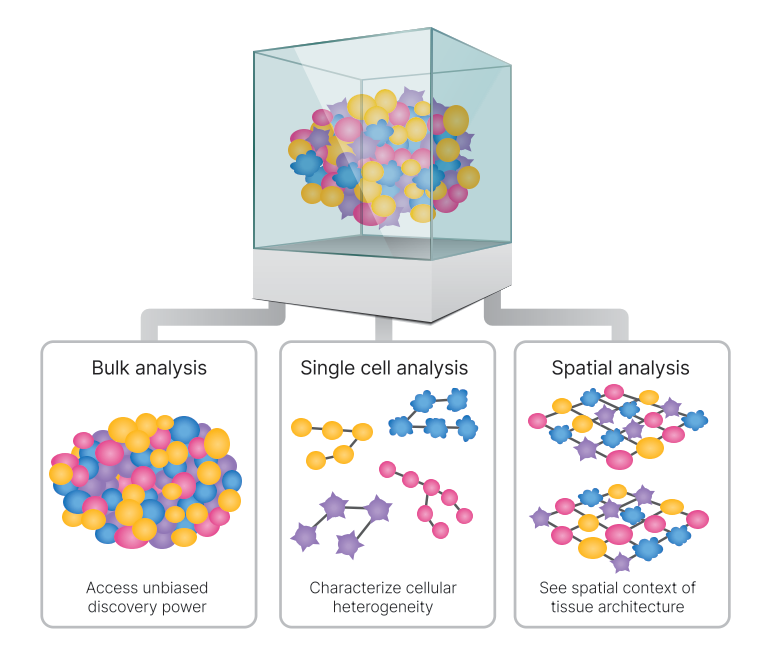
Bulk analysis
Single cell analysis
Single-cell sequencing is used to characterize hundreds to tens of thousands of individual cells from a tissue. This method reveals cellular heterogeneity and provides a more comprehensive understanding of tissue composition. S
- Spatial analysis
Spatial RNA-Seq provides a previously inaccessible view of the full transcriptome in morphological context. Spatial RNA-Seq methods that retain the precise location of biological molecules in tissue samples can further our understanding of mechanisms in health and disease.